自20世纪60—70年代凝聚态物理这一概念被广泛接受后,这一学科经历了飞速发展。凝聚态物理主要研究的是固态和液态物质的几何与电子结构,以及由此带来的声、光、电、磁、热等微观和宏观的物理现象。而化学学科发展至今,尤其在近20年,随着理论化学和化学表征手段的进步,研究人员开始逐渐意识到了化学反应并不仅仅是从反应物到产物这么简单的关系。反应体系的物质结构层次对化学反应的进程起到至关重要作用。人们逐渐开始重视化学反应的原位表征,并对揭示体系中不同层次的物质结构在反应条件下的动态变化进行探索。这些恰恰可以被看作是凝聚态化学研究的萌芽。物理与化学一直是相互交叉、相辅相成的两门自然科学。目前仍有很多凝聚态物理的新现象和新理论涌现出来。将这些新的物理现象和理论引入到化学研究当中是一个非常值得思考和探究问题。本文将对一些相对较晚出现的凝聚态物理概念(例如,表面等离激元极化子、拓扑绝缘体、准晶、局部微静电/磁场、光-物质相互作用、交变磁体等)及其在化学研究中的一些尝试进行简单介绍,旨在说明凝聚态物理研究前沿在化学研究中的应用前景,为推动传统化学研究进入凝聚态化学阶段提供一些思路,促进凝聚态化学这一学科的建设。
【关键词】凝聚态物理 /凝聚态化学/ 表面等离激元 / 拓扑绝缘体 / 局部微环境 / 光-物质相互作用
【作者信息】第一作者:贾然;通讯作者:闫文付
1 引言:从固体物理到凝聚态物理
凝聚态物理是现代物理学的重要分支,其理论体系融合了统计力学、量子场论和电磁学等基础框架。研究内容涵盖:(1)材料合成与结构表征;(2)物性测量与数据分析;(3)理论建模与物理机制阐述。在学科组织上,中国物理学会未设专门的凝聚态物理学分会,而是通过31个专业委员会实现更精细的学科划分,其中22个(占比超过70%)与凝聚态物理学密切相关。据不完全统计,2024年全球举办了至少63场聚焦凝聚态物理的国际学术会议与研讨会,反映出该领域在国内外物理学界的高度关注与广泛兴趣。
凝聚态物理学主要研究的是粒子间具有相互作用的多粒子体系的物理性质。广义而言,除理想气体外,现实物质世界中几乎所有体系中的粒子之间均存在着或强或弱的相互作用,因而均可归属为聚集态的研究范畴。然而,当前凝聚态物理学主要聚焦于固态和液态体系,即粒子间存在较强相互作用的系统。凝聚态物理学者通过实验与理论相结合的方法来观察、理解并预测这些体系的物理行为,为材料科学及相关技术的发展提供了重要支撑。
对物质凝聚态的探索,可追溯至19世纪英国化学家Humphry Davy与Michael Faraday对气-液相变的研究。在此基础上,爱尔兰化学家Thomas Andrews引入了“临界点”概念用以描述气态和液态共存且无法区分的状态;荷兰物理学家Johannes van der Waals则为其提供了坚实的理论模型。这一领域的决定性突破发生在1908年,苏格兰化学家James Dewar与荷兰实验物理学家Heike Kamerlingh Onnes分别实现了氦气的液化,开启了低温物理学的新纪元。氦气的成功液化,为Heike Kamerlingh Onnes在3年后(1911年)于4.16 K低温下发现汞的超导现象提供了关键的前置条件,使其成为凝聚态物理学史上里程碑事件。Onnes也因此荣获1913年的诺贝尔物理学奖。随后,作为超导材料的另一重要基本特性——抗磁性(即Meissner-Ochsenfeld效应),在1933年由德国物理学家Fritz Walther Meissner和Robert Ochsenfeld联手发现。
人类对磁现象的认知源远流长。早在公元前6世纪,古希腊哲学家泰勒斯便对磁石有所记载;而在中国,公元前4世纪的《鬼谷子》以及东汉王充的《论衡》中,亦有关于磁性的论述。然而,将磁性作为一种物理相变进行系统性科学研究则始于近代。法国科学家Pierre Curie的贡献是其中一个里程碑,他揭示了铁磁性物质在特定温度(后称“居里温度”)会转变为顺磁性的相变过程,并提出了居里定律(Curie law)。1907年,法国物理学家Pierre-Ernest Weiss在此基础上引入“磁畴”概念,并运用平均场理论修正了居里定理,提出了能更准确描述物质在高温下磁化率的居里-韦斯定律(Curie-Weiss law)。对磁性相变的微观描述,则由德国物理学家Wilhelm Lenz与Ernst Ising于1925年提出的伊辛模型(Ising model)所开创。该模型的精确解揭示了:一维周期性排列的磁偶极矩无法产生自发反铁磁-铁磁相变,而二维的方晶格模型则是能够展现铁磁-顺磁相变的最简单体系。同年,荷兰-美国理论物理学家George Uhlenbeck与Samuel Goudsmit合作发现了电子自旋(Electron spin)。这一发现从根本上揭示了物质磁性的量子起源,并由著名的Stern-Gerlach实验所直观证实。
随着19世纪末物理学的发展,一系列经典理论无法诠释的实验现象相继涌现,预示着一场深刻的物理学革命即将来临。英国物理学家Sir Joseph John Thomson通过对阴极射线的研究,不仅发现了电子的存在,更揭示了电荷的非连续性。紧接着,在20世纪的头一年,Max Planck为完美解释黑体辐射光谱,首次提出了能量量子化假说:谐振子发射的能量是其固有频率的整数倍,而联系二者的比例常数即“普朗克常数”。这一革命性的思想在1905年得到了Albert Einstein的进一步发展。Einstein将能量量子的概念推广至光,提出光量子(后称光子)假说,并以此为基础,成功地诠释了金属表面光-电效应现象。这些早期的、零散的量子思想,共同动摇了经典物理学的根基,为新的理论诞生准备了条件。
从“旧量子理论”向系统性理论的飞跃,始于 1924年。法国物理学家Louis Victor de Broglie公爵提出了“物质波假说”,将波粒二象性从光子推广到所有实物粒子,构成了新理论的转折点。同年,德国理论物理学家、数学家Max Born在工作中首次使用了“量子力学”这一术语,正式宣告了一个全新物理学分支的诞生。量子力学的建立,为人类理解原子、分子乃至宏观物质的微观行为提供了前所未有的强大理论武器。它不仅是研究除万有引力外所有基本相互作用的共同基础,其深刻的原理和强大的分析能力,更极大地促进了物质科学的研究,直接催生了以理解和调控多粒子集体行为为目标的凝聚态物理学。因此,对量子力学发展脉络的回顾,是理解凝聚态物理学理论根基的必要前提。
物质根据其内部粒子之间的相互作用的差异:可呈现为固态、液态、气态、等离子态、玻色-爱因斯坦凝聚态、费米子凝聚态等多种形态。其中,固态与液态因其粒子间存在不可忽略的强相互作用,统称为凝聚态。
在凝聚态物质中,固体材料根据其源自排列的有序性,又可分为3类:晶体(Crystal)、无定形体(Amorphous)和准晶(Quasicrystal)。晶体因其原子具有长程平移对称性、结构简洁明确,长期以来是固体物理理论研究的基石。无定形体则长程无序、结构复杂性虽为理论研究带来挑战,但其大量非化学计量配位数的活性位点,却为催化等化学应用提供了独特的优势。介于二者之间的是准晶—— 一种仅有长程取向序(例如5次、7次、8次、10次、12次旋转对称性)而无平移周期性的特殊固体形态[25]。准晶材料,多为合金,因其低导热导电、高硬度等特性,在功能涂层和材料增强等领域展现出应用潜力。
最初,对晶体学、冶金学、材料力学及光、电、磁性质的研究是相对独立的。直至20世纪40年代,这些主题才被系统地归入“固体物理学”。得益于半导体、超导、核磁共振等领域的重大突破,固体物理学为满足工业界的技术需求而飞速发展。然而,随着研究的深入,物理学家们逐渐认识到,液体、超流体等非固态体系与固体在物理规律上存在深刻的共性。“固体物理学”这一名称的局限性日益凸显。
这一概念上的演进,最终促使了“凝聚态物理学”这一更具包容性术语的确立。早在1962年,瑞士物理学家Georg Busch在西德创办了由Springer出版的物理学期刊Physik der Kondensierten Materie(《凝聚态物质物理学》)。1963年10月,美国物理学会下属的著名物理学期刊Physical Review发表声明,将在次年划分2个独立的专题部分,并在1970年最终被正式拆分为两个部分,其中一个主要致力于原子、分子和凝聚态的物理学研究。1978年4月,美国物理学会理事会议经过投票以15∶7的结果正式将其下属“固体物理分会”更名为“凝聚态物理分会”,标志着物理学界从概念上承认了适用于割裂凝聚态物质的普适性理论框架和研究范式。
凝聚态物理学的建立过程,本身就体现了物理与化学两大学科的深度交融。尽管通常认为化学构建于物理定律之上,但历史上,对凝聚态行为的早期关键探索(如相变、电化学等)多由化学家完成。物理学与化学实则相互依存、彼此支撑。在当代科学中,学科界限日趋模糊,重大科学问题的解决愈发依赖于交叉学科的视野。
化学研究的核心是化学反应—— 一个涉及物质转化和能量变化的动态过程。然而,任何化学反应都并非孤立发生,而是深植于其所在的凝聚态环境(如溶剂、催化剂、界面等)之中。传统的化学研究,包括其二级学科“固体化学”,往往更侧重于反应物与产物的静态结构与性质。但事实上,反应环境中的电场、磁场、应力场,以及溶剂分子、催化剂结构等,都在反应过程中发生着动态演变,并深刻影响着反应的路径、选择性与产率。异相催化就是一个典型例证:反应不仅发生在固态催化剂表面,更受到活性位点周围微环境的动态调控。这种复杂性,已无法再用传统的合成化学或催化化学等单一学科语言进行简单描述。
在此背景下,为推动化学学科在根本思想上的革新,徐如人先生高瞻远瞩地提出了建立“凝聚态化学学科”的学术思想,倡导在凝聚态的整体框架内审视化学问题。凝聚态化学的核心,是关注“多层次结构影响下的化学反应”,其重要分支——凝聚态结构化学——尤其应受到重视。在传统的结构化学教学和研究中所关注的重点主要聚集在孤立的分子和晶体的几何与电子构型,以及由此导致的物理表象(例如对各种图谱的理解)等方面。这就导致结构化学更像是研究“化学的结构”,甚至有低年级学生认为更偏向于物理学和数学。在凝聚态化学的框架下,化学研究的是反应及其动态过程,不再是孤立的静态体系。随着原位表征技术的长足发展,已经直观的证实了,催化剂表/界面活性位点周围的结构虽然不直接参与反应,但也会因为构型的改变令活性位点周围产生局部微环境物性动态变化,从而影响到化学反应进程。因此,凝聚态结构化学研究的落足点应该在于:在反应条件下,反应体系物质结构所发生的动态演变过程及其对化学反应选择性和最终产率的影响。
传统结构化学的研究重点多集中于孤立分子或理想晶体的静态几何与电子构型。而凝聚态结构化学则将焦点转移至动态的、多层次的反应体系。它强调,在真实反应条件下,催化剂表界面、溶剂壳层等非直接参与反应的结构,其动态演变所引发的局部微环境物性变化,对化学反应进程起着决定性作用。
因此,凝聚态结构化学的研究落脚点在于:在真实反应条件下,体系物质结构的多层次动态演变过程,及其对化学反应选择性与产率的调控机制。
与传统结构化学相比,其核心区别体现在3个维度:(1) 结构的多层次性:从原子、分子到介观、宏观,关注跨尺度的结构关联;(2) 过程动态演化性:借助原位表征等手段,研究结构在反应过程中的实时变化;(3) 环境的决定性影响:强调微环境(场、溶剂、界面等)对反应行为的核心调控作用。
本文旨在以固态体系中的化学反应为例,介绍若干典型的凝聚态结构因素,并对凝聚态物理学中的新近物理现象在化学领域的应用前景进行展望,以期阐明凝聚态结构化学的学科要义与重要性,为这一新兴交叉学科的发展奠定概念基础。
2 化学反应体系中的凝聚态
凝聚态物理学经过几十年的发展,获得了飞速的进步和令人瞩目的成就。量子力学作为凝聚态物理的基石,为理解物质在微观尺度上的行为提供了有力的理论工具。然而,当试图将这些源于理想化模型(如孤立原子、分子或完美晶体)的物理基础理论直接应用于复杂的化学反应体系时,其适用性和预测能力往往会面临严峻挑战。这并非理论本身的“失败”,而是源于凝聚态物理与凝聚态化学在研究对象和核心范式上的根本差异。
凝聚态物理的理论优势在于处理高度有序或者可通过微扰理论修正的体系,其研究范式倾向于“化繁为简”——通过抽象和简化,剥离次要因素,以抓住支配体系行为的核心物理规律。与此不同,绝大多数化学反应并不是发生在如此纯粹的体系当中。化学反应本质上是一个动态的、多因素耦合的复杂系统,其中,物态、相界面、溶剂、分散剂乃至环境中的局域场,都作为活性参与者,共同决定着反应的进程与走向。
这种差异根植于两者研究目标上的区别:物理学旨在揭示普适的自然法则,而化学则致力于创造和调控性物质。因此,化学家在研究中更侧重于“化简为繁”——主动引入和利用多种可控变量(如催化剂、添加剂、溶剂体系、外场等),以期对反应路径、速率和选择性进行精准的人为干预。
正是基于对化学反应复杂性的深刻认识和对精准调控的追求,凝聚态结构化学思想应运而生。秉承这一核心思想,本章将通过具体实例,系统剖析界面效应、静电/磁场、活性位点微环境以及光场(电-磁交变场)等关键凝聚态因素,是如何在化学反应过程中发挥其动态调控作用的。
2.1 反应中的动态界面结构
根据物质的聚集状态,界面化学反应可大致分为6种类型:固-气、固-液、固-固、液-液、液-气、气-气。考虑到凝聚态化学的研究范畴和本专辑的范围,本文将聚焦于前3类,即以固态物质为核心的表/界面反应体系。固态表/界面在自然界与人工系统中普遍存在,其结构复杂性是理解其化学行为的基础。这种复杂性不仅体现在原子、分子的微观排列上,更体现在由不同热力学条件决定的“相(Phase)”这一更高层次的结构上。例如,仅SiO2一种化合物,目前已经证实的晶相就多达14种,包括其二维形态。固体材料的晶相结构,在很大程度上决定了其本征的物理和化学性质。
然而,传统上对固体表/界面结构的静态认知正在被颠覆。越来越多的证据表明,固态表/界面远非化学反应中一个静默的旁观者,而是会随着化学反应的进程而发生深刻的动态演化。在典型的催化反应中,这些动态演化可表现为多个层面:催化剂的宏观形貌与纳米颗粒尺寸的改变、反应活性位点配位数与电子结构的调整,乃至载体或基底材料结构的重构等。这些演化过程依据其能否在反应停止后恢复原状,可进一步区分为可逆与不可逆两类。图1以电催化析氢反应为例,展示了几种典型可逆与不可逆动态表面结构演化方式。
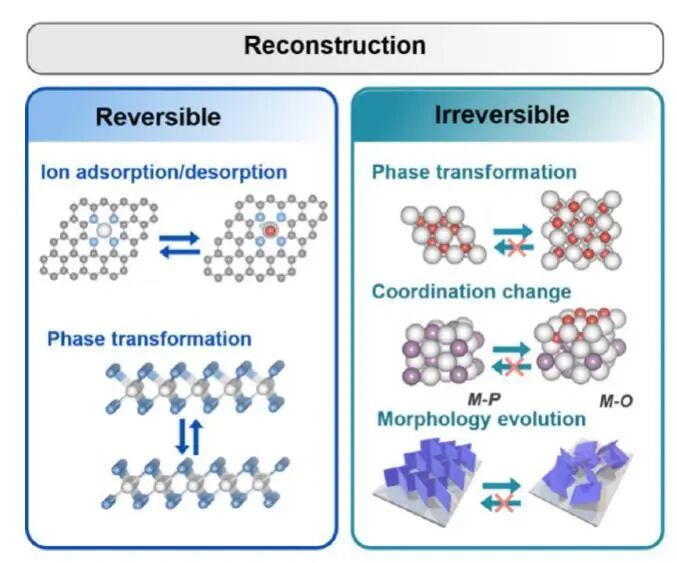
图1 电催化析氢反应(HER)过程中可能出现的表面结构可逆与不可逆动态演化过程示例图
2.1.1 固-气界面反应
在固-气界面发生的第一步应该就是气体分子在固体表面的吸附。根据固体表面和气体分子性质的不同,吸附行为主要可以分为:物理吸附、化学吸附以及介于二者之间的物理-化学吸附。在界面化学反应的研究中,通常不考虑吸附能较弱的物理吸附。理论研究早已指出,晶体表面从体相中暴露出来后,表面原子层会首先发生弛豫。这种弛豫现象在配位程度较低的位点(例如台阶、缺陷处)尤为显著。此外,当表面发生较强的化学吸附时,吸附物种会对吸附位点附近的原子产生显著的的诱导效应,从而引起表面结构的重整。这种结构变化的发生时间通常与相应的化学过程时间尺度相符。实验研究早在20世纪90年代便已揭示,表面的重构能力与反应活性之间存在密切关联。1996年,有实验研究明确地观测到在Pt(111)表面吸附S原子并在600 ℃进行30 s退火后会形成如图2a所示的双原子厚度的Pt阶梯状构型。然而,当在室温条件下Pt(111)表面发生S-CO共吸附时会出现结构重整,如图2b所示,Pt阶梯的高度会降低为单原子厚度,且S与CO交替吸附在Pt台阶面上。有趣的是,重新升温到600 ℃后,Pt(111)表面重新恢复到原来双层台阶的状态。这一研究给出了化学吸附物与吸附剂表面构型之间相互关联的直观图像。随着实验技术的发展,同样是对于Pt表面吸附CO分子后构型重整的研究,这个研究组在14年后又报道了Pt(557)表面原有的周期性台阶结构随CO分子覆盖度变化发生明显重构的现象。在图2c中可以看到在超真空条件下存在的Pt(557)表面完美的阶梯状表面结构。当体系中通入少量CO气体时,表面结构已经开始出现较明显的变化,阶梯数量变少,出现波浪形结构,如图2d中所示。当CO气体压强升高到1 torr后,如图2e, f所示,其表面不再呈现阶梯状,出现尺寸约为2.2 nm的三角形Pt纳米颗粒。作者表示,这种表面破碎成纳米团簇的结构重整行为是为了释放相邻CO分子间的排斥作用,降低其表面能。值得一提的是,这项研究中还提到,同样条件下,在Pt(332)表面观察到的结构重整结果并不相同。这些研究结果清晰表明,固-气界面的结构很容易受到气体成分、浓度、反应类型等因素的诱导,在反应过程中呈现持续的动态演化。越来越多的实验证据为这一结论提供了强有力的支撑。
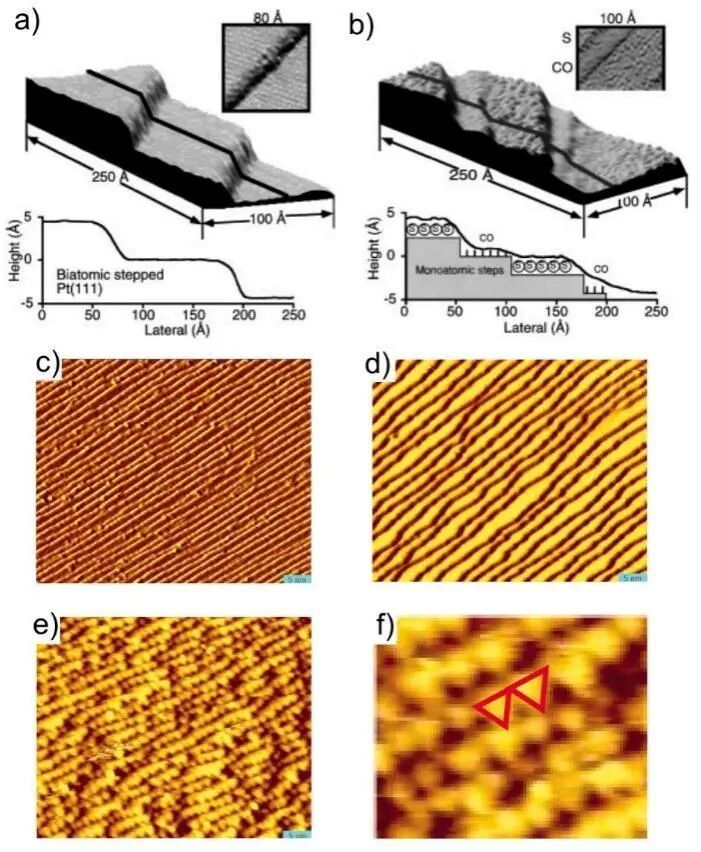
图2 (a) Pt(111)表面吸附S原子并退火至600 ℃后STM形貌图;(b) 室温条件下Pt(111)表面发生S-CO共吸附时的STM形貌图;(c) 超真空条件下Pt(557)表面STM形貌图;(d) 通入5×10-8 torr CO气体后Pt(557)表面形貌;(e) 通入1 torr CO气体后Pt(557)表面出现三角形纳米颗粒;(f) 1 torr CO氛围下放大的Pt(557)表面形貌
石墨烯无疑是21世纪前20年最具热度的研究领域之一。在过渡金属表面通过气相沉积法(CVD)合成石墨烯的过程中,可以观测到金属-碳氢化合物气体界面结构动态演化。过渡金属表面与碳原子的相互作用在石墨烯热潮兴起之前便已备受关注。在高温条件下,碳原子可以溶解进入过渡金属内部,形成固溶体。根据碳原子在过渡金属内的溶解度,CVD合成石墨烯可分为面分解生长(Decomposition)和表面偏析生长(Segregation)2种类型,分别以Cu和Ni为典型催化金属。在1000 ℃高温条件下,碳原子在Cu表面的溶解度约为0.04%(原子分数),而在Ni表面则高达2.03%(原子分数)。
在Cu(111)表面,碳氢化合物气体分子(通常为甲烷、乙烷、甲醇等)作为碳源被催化分解脱氢,释放出的碳原子在表面扩散、成核并生长,最终形成高质量的单层石墨烯。与之形成鲜明对比的是Ni表面的石墨烯合成过程。由于Ni对碳具有较高溶解度,高温下分解的碳原子可渗入Ni内部,形成亚稳态的Ni-C固溶体。当温度降低后,碳原子在Ni金属体中的溶解度降低,进而发生偏析,驱动石墨烯在Ni(111)表面的外延生长。图3中的计算机动力学模拟结果直观的给出了Ni3C晶体中C元素的析出和表面石墨烯成核过程。可以看出,随着C原子的不断析出,部分Ni原子也会被拉出表面,为晶体内层更深处的C原子外迁提空间。Ni原子与表面析出的C原子通过σ键形成Ni-Cn-Ni桥联结构,将聚炔链稳定在Ni表面。理论研究进一步指出,表面C网络的形成主要归因于Ni原子的迁移性。这种金属-气体界面在高温反应过程中的动态结构变化,正是凝聚态结构化学研究的核心问题。
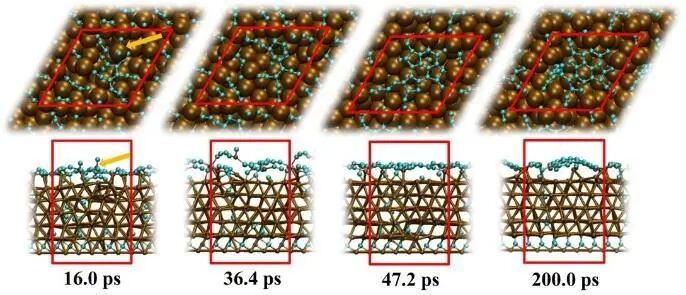
图3 Ni3C晶体表面C元素析出的QM/MD模拟瞬时结构截图,其中棕色和青色小球分别代表Ni原子和C原子
2.1.2 固-液界面反应
固-液界面在自然界和技术领域中极为常见,其上发生的化学反应广为人知,例如金属在盐溶液中发生的腐蚀、食盐晶体表面的溶解、离子晶体在过饱和溶液中的结晶析出,以及电解质溶液中的电催化反应等。这些常见的化学反应均伴随着界面结构的动态演化,而非静态过程。固-液界面结构的动态变化显著影响反应进程。由于固-液界面附近液体具有较强的长程迁移性,液相内部靠近界面的结构始终处于动态变化之中,这一现象显而易见。因此,本文重点探讨固相侧,尤其是与液相接触的固体表面的动态行为,而液相结构的动态变化归因于界面微环境因素,并将在后面简要讨论。
腐蚀是材料表面与环境相互作用而发生劣化的现象,常见于暴露在水溶液中的金属表面。这一复杂的电化学反应过程会导致金属表面形貌和化学组成的动态变化。以碳钢在NaCl溶液中的局部腐蚀(点蚀)为例,其过程通常分为3个阶段:诱导期、扩展期和饱和期(Induction, propagation, and saturation periods)。界面处Cl-的存在会首先破坏金属表面氧化膜,产生缺陷位点,并引发后继的反应。在扩展期,腐蚀点的生长速率会显著加快;当蚀坑广泛分布时,腐蚀速率趋于饱和,随后因表面钝化和扩散限制,点蚀生长速率随时间降低。金属材料表面固有的缺陷、表面应力以及溶液中的化学成分均显著影响腐蚀过程。另外,腐蚀产物形成的腐蚀层也会动态的改变界面的形貌,进一步影响腐蚀速率。在腐蚀过程中,金属阳离子会通过水凝胶层扩散,并与金属氢氧化物的沉淀耦合,产生由溶液反馈过程所引起的化学波(Chemical wave),从而形成周期性聚集的腐蚀产物图案。
晶体与其溶液之间形成连续的动态界面,溶质分子在晶体表面不断附着或脱附,驱动晶体的生长或溶解。晶体生长的机制包括台阶流、螺旋生长、二维成核等,高度依赖于溶液的过饱和度。在低过饱和度下,晶体表面呈现台阶状推进生长;而在较高的过饱和度条件下,晶体表面易产生位错,从而诱导螺旋生长和二维核。作为晶体生长的逆过程,晶体溶解具有非线性特征,其速率在时间和空间上均存在依赖性。晶体表面的形貌,尤其是缺陷,会引发初始快速的溶解;随着较稳定的次表层暴露,表面活性降低,溶解速度减缓并趋于稳定。晶体溶解速率还强烈依赖于界面处溶液部分的动态饱和度。台阶波(Stepwaves)模型解释了脉冲式物质流以及晶格缺陷在驱动溶解中的关键作用。
许多催化反应发生在固-液界面,固体催化剂在液相中通常会经历表面结构和组分的动态变化,显著影响催化活性和反应选择性。因此,解析界面结构动态演化过程是确定催化剂表面活性结构的关键。在电催化分解水析氧(OER)反应中,常选用金属、金属氧化物或硫化物作为催化剂,但其表面发生重构后形成的氧化物、氢氧化物、羟基氧化物才是真正的活性位点。在反应过程中,固体催化剂中某些特定元素的迁移驱动表面重构,并由此改变其催化性能。以SrIrO3钙钛矿材料电催化OER反应为例,如图4a~c所示,其中的Sr²⁺会随时间迁移(溶解)至溶剂当中,O2-引起表面重构,形成无定形层。以NiCo双金属氟化物为电催化剂的OER反应中,F元素的迁移导致其表面重构, 产生更多的表面活性位点,增加了电化学活性表面积。铁可以在很大程度上提升Ni(氢)氧化物在OER反应中的催化活性。当电解质中存在Fe³⁺阳离子时,NiO电催化剂的表面会与之结合形成层状NiFe双氢氧化物,如图4d所示。然而,随反应时间延长,FeOx在表面的额外聚集会降低催化活性。
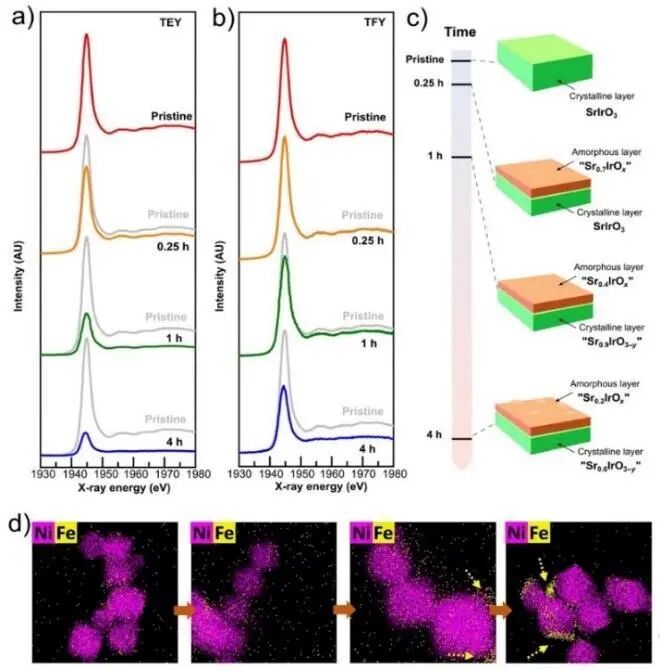
图4 (a) SrIrO3钙钛矿中Sr元素L3边X射线吸收近边光谱(XANES)在总电子产额TEF和(b) 总荧光产额TFY中随反应时间的变化;(c) 按照XANES谱线获得的随时间变化Sr元素浓度的直观图像;(d) 1 mmol/L硝酸铁溶液中获得的NiO晶体Ni/Fe化学X射线能谱(EDX)随OER循环次数增加的变化
当前,学术界已认识到,理解催化剂材料的结构,尤其是结构、组分与表面重构之间的关系对于设计高效催化反应体系至关重要。掌握界面处可能存在的瞬态非晶相、缺陷等结构特征,阐明表面活性位点动态的形成和演化机制,对于异相催化反应具有重要的意义。
2.1.3 固-固界面反应
固态界面处的化学反应是烧结、扩散键合、相变、腐蚀等多种现象的基础,对设计高性能功能材料、提升技术效率以及增强材料耐久性具有重要意义。理解并调控固态界面反应是材料科学研究的关键课题。固态界面化学反应通常涉及两种或多种固态物质在接触界面发生化学反应,其中机械化学反应是典型代表。根据国际纯粹与应用化学联合会(IUPAC)的定义:机械化学反应就是直接由吸收机械能而引发的反应。常见的方式包括研磨、球磨和超声波处理,通过施加冲击、剪切或摩擦,为反应物提供克服活化能垒所需的能量,从而促进化学转化的发生。早在1820年,Michael Faraday通过“干法”研磨AgCl与Zn混合物,成功置换出Ag单质,验证了机械能诱导化学反应的可行性。
机械能促进固态界面反应的机制主要包括以下方面:(1)减少反应物粒径,增大比表面积,提高反应物之间接触概率,从而提升反应速率;(2)引入晶格缺陷,提高位点能量,形成高活性反应位点,提高整体反应活性;(3)诱导晶体反应物表面非晶化,降低反应能垒,进一步提升反应活性。如图5a所示,机械化学反应过程,反应物在机械能作用下经历弹性形变,进入非弹性形变状态(如相变、非晶化、缺陷生成),最终被活化,克服反应能垒,发生化学反应,包括共价键的断裂和形成,界面处质子或电子转移,以及单体聚合或分解反应的加速等。对于分子晶体,反应可分为固体内部和固体间两种类型(图5b)。此外,有机分子晶体与无机离子晶体通过机械研磨形成固溶体、超分子晶体或共晶。如图5c所示,机械研磨有机金属两性离子晶体和碱金属晶体,促使碱金属阳离子和卤素阴离子在机械能的作用下嵌入到[CoIII(η⁵‐C5H4COOH)(η⁵‐C5H4COO)]分子间,形成超分子配合物晶体结构。从本质上看,机械化学过程与凝聚态结构化学思想高度契合,其反应进程由动态演化的物结构驱动和调控,体现了结构与功能的内在关联。
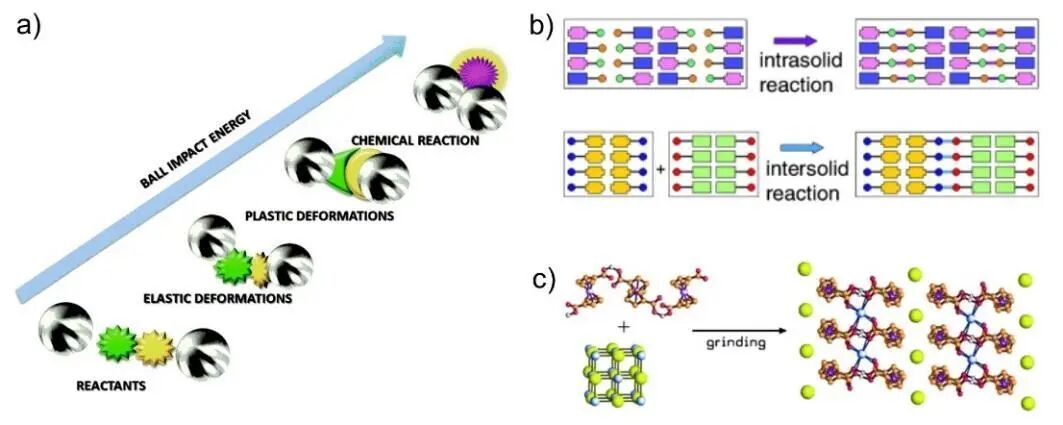
图5 (a) 机械化学反应示意图; (b) 分子晶体内部与分子晶体间发生的固态反应; (c) 研磨有机金属与碱金属卤盐MX晶体形成的超分子配合物
全固态电池因其优异的性能与安全性,被认为是实现高性能电池目标的理想体系。其核心特征在于采用固态电解质替代传统液态电解质。如图6所示,全固态电池中包含多种固-固界面,包括金属阳极-电解质、电解质-电解质、阴极-电解质以及导电添加剂-电解质等。化学反应主要发生于以下四类界面:阴极-电解质、阳极-电解质、导电填料-电解质、阴极积电器-电解质。界面电阻是制约全固态电池性能的关键因素,而界面电阻与界面处复杂的化学反应密切相关,因此深入研究这些界面化学反应对优化电池性能至关重要。
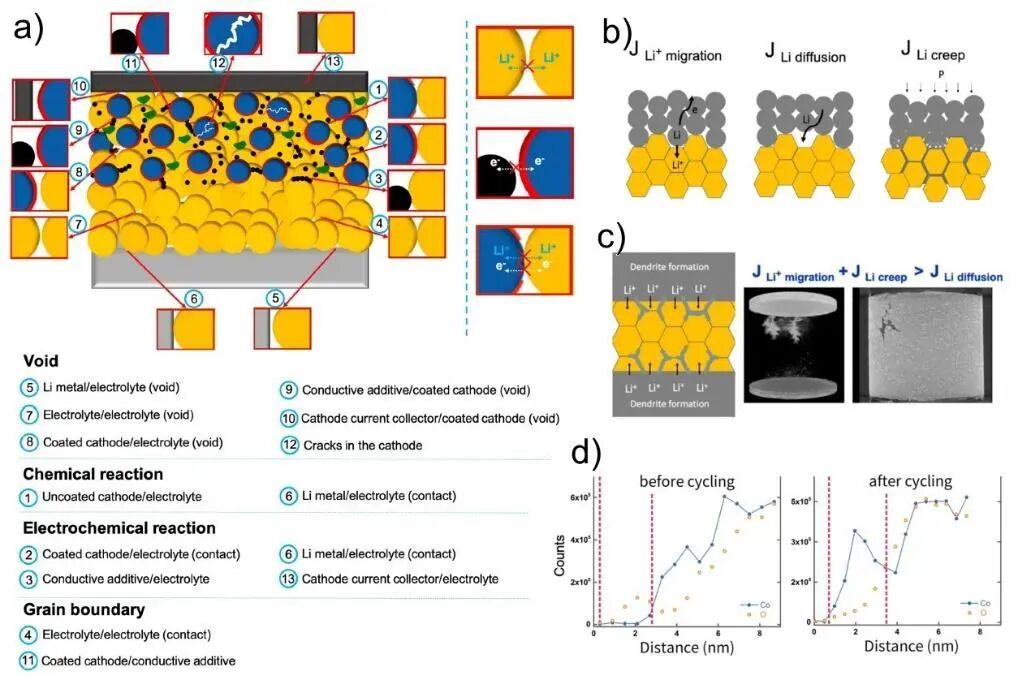
图6 (a) 全固态锂离子电池界面现象结构示意图;(b) 金属阳极-电解质界面处锂离子迁移方式;(c) 界面堆积压25 MPa时出现显著的Li枝晶生长状态;(d) 电池300次循环前后Co元素渗透入阴极包覆膜内,其中蓝色与黄色符号分别代表Co、O元素电子能量损失谱线扫描信号
以锂金属阳极与固态电解质界面为例,如图6b所示,界面状态(包括结构状态和应力状态)差异显著影响锂元素的迁移行为。不同的迁移行为进一步导致电解质中枝晶生长的程度各异(图6c)。类似地,阴极材料在充-放电循环中也经历动态结构演化。如图6d所示,在300次循环后,c-LiCoO2阴极材料中的Co元素明显向阴极包裹层扩散,反映了界面化学的动态变化。
在异相催化反应中,固-固界面常作为催化活性位点,其结构在纳米甚至原子尺度的精准调控直接影响催化性能,是大规模工业催化过程的核心。固态异质结催化剂的结构与组成决定了体系催化活性和反应机理。以Pt纳米颗粒催化CO生成CO2为例(图7),当Pt纳米颗粒置于惰性材料表面时,低温下,Pt纳米颗粒表面被CO分子覆盖,难以活化O2,导致无法有效催化CO氧化反应。为了同时活化CO和O2,通常需将温度提升至200 ℃以上,以诱导吸附在Pt纳米颗粒上的CO分子部分脱附,为O2吸附和活化提供位点。然而,如果将衬底材料替换为具有活化O2分子能力的CeO2晶体,Pt纳米颗粒与CeO2晶体表面可以分别负责活化CO和O2分子,从而显著提升低温催化能力。通过调控异质结界面结构,可有效控制肖特基能垒的形成与强度,促进不同种类载流子的分离,并可能增强压电催化能力。
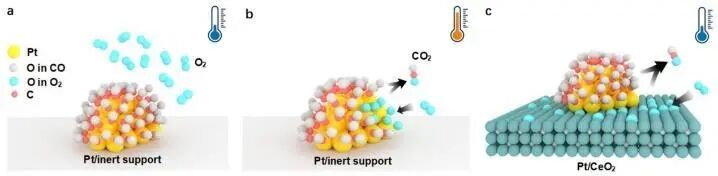
图7 Pt纳米颗粒催化CO氧化生成CO2反应:(a)低温情况下,Pt纳米颗粒置于惰性衬底;(b)高温情况下,Pt纳米颗粒置于惰性衬底;(c)低温情况下,Pt纳米颗粒置于活性CeO2表面
固态异质结催化剂界面在反应过程中会发生动态结构演化。以Cu与其他金属组成的界面为例,其在电催化CO2还原(CO2R)反应中可显著提高生成多碳化合物的选择性。Au-Cu双金属体系(Cu表面外延生长Au纳米颗粒)是研究电催化CO2R生成多碳醇的典型模型。根据传统理论,Au表面首先将CO2还原为*CO,形成高浓度的*CO并引发二聚化,随后在Cu表面生成高价值目标产物。然而,能量色散X光谱(EDS)分析表明,Au-Cu界面结构并非静态固定。反应过程中,A界面附近持续产生可动态迁移的Cu空位(Vacancy)和Cu离子,导致初始清晰的双金属界面逐步演化成为由Au-Cu合金支撑的Au@Cu核壳纳米团簇结构。具体而言,Cu(0)首先被氧化为Cu(I)和Cu(II),并在表面晶格中引入O元素,破坏局部原子构型。在偏压的作用下,Cu(I)和Cu(II)被还原为Cu(0),进而在附近产生原子空位。这些空位促进Cu和Au原子的互相扩散,形成Au-Cu合金层。由于Au纳米团簇表面较低的吸附能,电解质中的Cu离子容易沉积其上,形成Au@Cu核壳结构。这一动态重构由Cu(0)的氧化/还原过程驱动,颠覆了传统理论对反应机理的理解:被包裹在Cu壳中的Au团簇难以直接活化CO2分子生成*CO。更合理的机理是:*CO在Au-Cu合金层上生成,随后发生脱附并转移到团簇区域的Cu壳上,最终在Cu壳上转化为乙醇。这一修正与凝聚态结构化学的核心思想相符,即结构动态演化决定反应进程。类似地,另一项针对Cu68Ag32纳米线电催化CO2R的研究也观察到表面动态结构演化和合金层的形成。
近期研究表明,电催化CO2R反应生产高附加值多碳醇类产物时,可以利用稀土元素Pr独特的4f电子轨道和未占据的5d轨道调控相邻Cu原子的电子密度分布。通过Pr-O-Cu键的协同作用,可动态调控Cu元素的价态在(0~+2价),并形成自修复的稳定异质结界面。研究人员采用原位X射线发射谱(XES)分析方法揭示了反应过程中Cu元素价态的逐步变化。这些发现进一步印证了凝聚态结构化学在催化研究中的重要性。
随着研究技术的进步,凝聚态结构化学的证据不断丰富。当前,研究反应条件下结构动态演化的主要方法包括理论模拟和原位表征(In-situ characterizations)。理论模拟通过简化反应条件,预测和解析短时间尺度内反应体系的微观几何与电子结构演化。随着计算能力的提升,研究已从静态结构模拟转向动态反应过程的深入分析,力求贴近实验条件。以Au/TiO2纳米体系在700 K下的第一性原理动力学模拟(AIMD)为例,如图8所示,结果显示TiO2间隙缺陷Ti会倾向于向Au团簇内迁移,而非在表面析出并被O2氧化生长出新的TiO2岛。然后,理论模拟受限于计算能力,模型规模和模拟时间较小,难以全面反映复杂体系的动态行为。为了克服这些困难,理论学者不断尝试开发了多种新方法,如基于密度泛函的紧束缚方法(DFTB)、量子力学结合经典力学的方法(QM/MM)、反应力场(ReaxFF)、机器学习(ML)等,期待未来计算硬件与软件的突破,尤其是AI4Science取得突破,实现更接近真实条件的化学反应模拟,从而帮助人们在根本上对化学反应进行理解和设计。
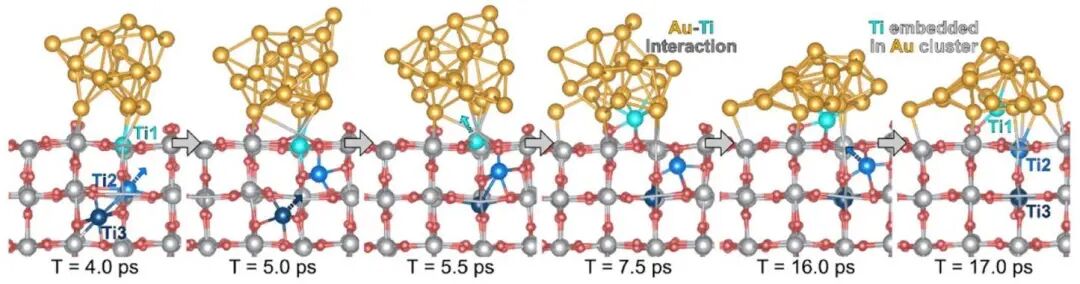
图8 第一性原理动力学(AIMD)模拟700 K条件下TiO2表面Ti原子扩散进入Au20团簇的过程
原位表征技术的发展为实时检测反应过程中的动态结构演化提供了有力工具,尤其是工况原位表征(Operando characterizations)技术,通过结合结构表征与催化活性以及选择性测量,显著提升了分析精度。原位表征技术可分为光学技术和光谱技术两大类:原位光学技术直接揭示催化剂在反应过程中几何构型的演化,原位光谱技术通过分析价态、配位数等变化来间接地获得结构演化信息。然而,当前原位表征技术仍面临局限性,如对于一些辐照、高温熔融、超高压、强磁场等较苛刻的条件下的反应体系难以适用。此外,信号采集速度、精度以及数据处理的复杂性等限制了其广泛应用。为克服这些挑战,研究者致力于开发更加先进的原位表征技术,并通过多技术联合应用以期更全面地解析化学反应过程。
理论模拟与原位表征的进步表明,异相催化领域已经深刻认识到动态结构演化对催化性能的决定性影响。因此,将这一研究视角融入更广泛的化学领域,强调凝聚态化学和凝聚态结构化学的理论框架,不仅具有重要意义,也正当其时。
2.2 外电场
电场是物理学中的基本概念。在其作用下,凝聚态体系常表现出独特的物理行为与化学现象。在电池和电催化等化学体系中,外电场的影响尤为显著。从微观角度看,化学反应位点因周围物质组成和构型的动态变化,处于瞬时、动态的介电环境中,受到反应电场、内建电场、诱导电场或压电场的作用。本文聚焦外电场对化学反应的影响,将微观尺度瞬时电场讨论纳入后续的微环境一节中。
早在20世纪30年代,挪威裔美国物理化学和理论物理学家Lars Onsager通过扩散方程描述了初始分离的离子对在库仑相互作用和外电场的影响下发生复合的数学模型,推导出离子对逃逸复合的概率表达式。后续研究发现,在高场强外电场作用下,电解质中离子对表现出逆温效应,即逃逸复合概率随温度升高而降低,为凝聚态化学研究提供了早期理论启示。近年来,外电场作为化学反应的新型调控手段重新受到关注。外电场的强度、方向、频率等参数直接影响到凝聚态系统中的分子构型、几何排布以及电子结构,从而调控反应活性,促进、抑制或诱导化学反应。典型的例证是沿特定方向施加的外电场可改变化学键的性质,甚至引发化学键的断裂和重构。若外电场方向与电子在反应物向产物转化过程中的重组的方向(反应轴)一致或相反,可分别催化或抑制反应,并可能引发从协同反应机制到逐步反应机制的转变。此外,通过增强电荷分离共振,外电场可稳定共价化合物的分子结构甚至过渡态结构,显著降低反应能垒。隧道扫描显微镜(STM)探针尖端所施加定向静电场可加速Diels-Alder反应,首次实验验证了外电场调控非氧化还原键的形成,直接证明了通过外电场操控化学反应动力学和热力学的可行性,为多相催化方法提供了新的思路。近期的研究发现通过STM技术控制的外电场可以提高Diels-Alder反应速度3倍以上,但对逆反应速度却没有影响,显示出电场对反应选择性进行操纵的潜力。除此之外,同样的STM控制的外电场还可被用来进行电子催化单分子脱氢反应。研究人员通过STM中电极间电场驱动电子转移,从而发生电子催化的1,2-二(4-吡啶鎓)乙烷(DPA2+)生成1,2-二(4-吡啶鎓)乙烯(DPE2+)的单分子拖行反应(图9)。
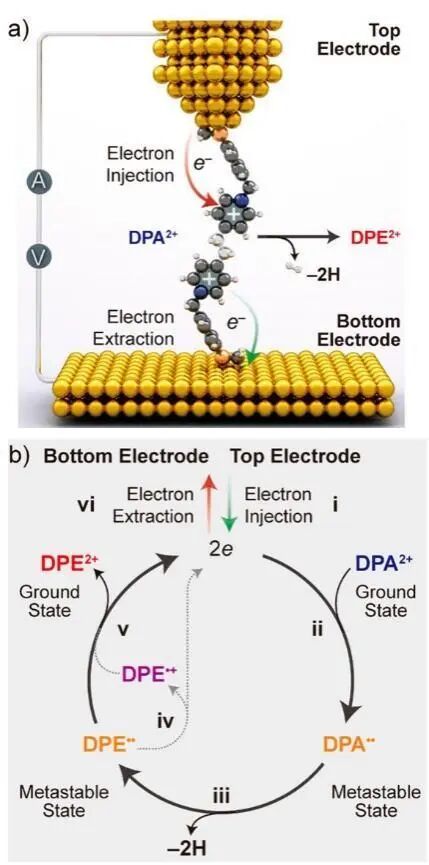
图9 STM电极间电场促进电子转移导致的DPA2+单分子脱氢生成DPE2+反应:(a) 反应装置示意图,(b) 脱氢反应路线图
从量子力学视角,外电场通过诱导轨道杂化调控非氧化还原化学键的生成。以氢原子为例,主量子数n=2时的4个简并的能级在无外场时因对称性正交,无法杂化。然而,沿z轴施加外电场会引发Stark效应,形成能量分裂的2s±2pz杂化轨道,如图10所示。这一机制可打破轨道杂化的对称性禁阻,使违反Woodward-Hoffmann规则的反应或正面亲核置换反应(Front-side nucleophilic displacement reaction)成为可能。外电场还可通过稳定特定自旋态的过渡态调控反应自旋选择性,其方向与施加方式是控制立体选择性和反应性的关键因素。因此,外电场作为一种通用工具,在精细化学合成中具有广阔的应用潜力。
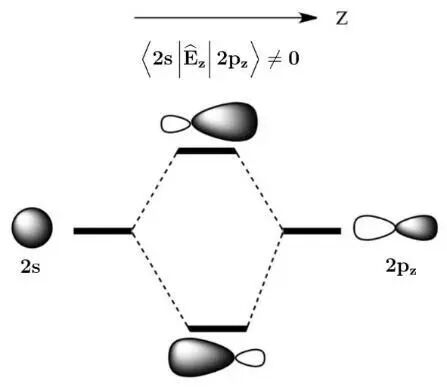
图10 在电场算符Êz 作用下氢原子2s与2pz轨道杂化示意图
在光催化体系中,外电场促进正/负电荷(载流子)分离尤为重要,是实现高效太阳能转换的关键策略之一。光催化过程中,光生载流子的复合会降低光子能量利用率以及催化效率。施加恰当的外电场可以灵活的调节半导体材料的电子结构,诱导电偶极子极化,改变局部几何构型或引入局部应力,从而通过能带间隙以及带边位置的变化影响光子吸收、价电子的激发以及反应活性。如图11所示,二维范德华异质结光催化剂在外电场作用下,其能带间隙和带边位置发生显著变化。此外,外电场可改变半导体材料的光学吸收边并导致其折射率出现相应的变化,在强外电场作用下甚至引发Franz-Keldysh效应,使电子吸收低于带隙能量的光子而被激发,尽管该效应需要高电压(数百伏特)在化学反应中较少见。对于具有明显电偶极矩的分子体系(如液态体系),外电场可改变体系相变临界温度或诱导链状自组装行为。然而,外电场的定义需要考虑实际反应装置的介电环境,不同介电条件可能导致体系行为截然相反。
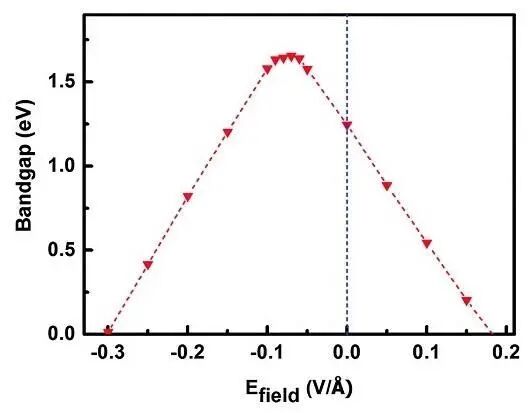
图11 采用PBE泛函进行计算机模拟所获得的MoS2/PbI2范德华异质结体系能带间隙与外电场强度之间的变化规律
近期电化学研究表明,脉冲电场通过周期性调控催化或抑制体系内电荷分布,可显著提升反应效率和选择性。例如,通过动态调控非对称电荷分布,可提高C-N偶联效率以及二氧化碳还原反应的效率。外电场在化学反应中的多维调控潜力,为凝聚态化学研究提供了新的理论与实验方向。
2.3 其他外场
除外电场外,化学反应调控中常涉及外磁场、温度梯度场、应力场以及电磁场(光场)等多种外场。通常大多数外场以矢量场的形式沿特定方向施加,与温度、压力等标量控制因素形成鲜明对比,且在实际反应中常以耦合形式协同影响反应进程。本节聚焦单一的外磁场、温度梯度场、应力场以及电磁场对化学反应的调控作用,通过典型案例进行简要分析。
尽管磁相互作用较库仑作用低几个数量级,通常在化学反应能量平衡中被忽略,但磁微扰所带来的近简并势能面交叉对于反应速率、产率以及光谱特性具有重要影响。在涉及电子自旋的反应中(如单线态和三线态转换,尤其是在过渡金属体系中),局部非均匀的磁场可诱导自旋态相位的交换,调控反应过程。由于电子自旋的磁矩远大于质子磁矩,非均匀磁场作用下,二者的进动频率差异显著,从而影响长寿命的激发态自由基或自由基对的反应选择性。外磁场通过调控核-电子自旋共振中的自旋态布居,改变自由基对的角动量,进而实现磁催化效应。硅同位素中具有核自旋为1/2的29Si因磁同位素效应,其氧化速率约为非磁同位素的两倍以上,而非磁性Si氧化的速度之间几乎没有区别。通二次离子质谱法(Secondary ion mass spectrometry)分析进一步证实,外磁场可加速固态Si晶体氧化。此外,外磁场强度对反应产率的影响显著。如图12所示,在Pd@Co3[Co(CN)6]2催化Suzuki偶联反应(溴苯和苯基硼酸生成二苯)中,外磁场增强产率,但其效果随温度升高(至60 ℃)显著减弱,因顺磁性Pd纳米颗粒的磁化率会随着温度的升高而降低。
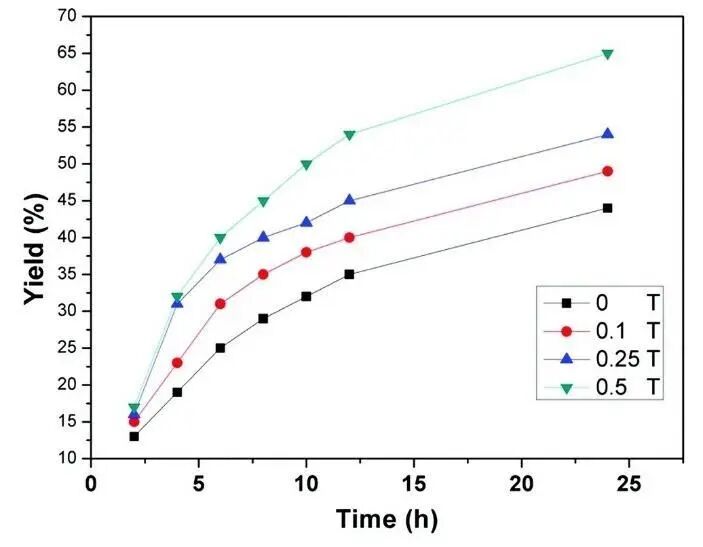
图12 外磁场增强Pd@Co3[Co(CN)6]2纳米颗粒催化Suzuki偶联反应合成二苯产率
温度作为化学反应调控的关键因素,在异相反应体系中因反应位点的相对集中形成显著的温度梯度场。人为调控温度梯度可通过移行溶剂浮区法、热梯度化学气相渗透或温度梯度胶体电泳等技术实现。然而,温度梯度可能导致反应速率不均、过度反应或应力分布不均匀等不利影响,同时也可作为稳定反应的驱动力,调控扩散通量以及反应空间定位。在流体体系中,温度梯度通过自由能转移驱动萃取或非平衡合成。在非平衡稳态体系(如生物细胞代谢或太阳能电池),温度梯度维持净通量,调控动力学行为。例如,钙钛矿材料在光照下因导热率差异形成局部温度梯度,导致化学不稳定性增强,即使温度远低于热分解阈值,温度梯度仍可诱导外源物种扩散,加速分解过程。早期Varshni、O’Donnell等,以及后来Lee等对温度与半导体能带关系先后进行的理论研究表明,半导体能带间隙的变化不仅与温度有关,还受应变及热膨胀系数影响。在气-液-固三相生长纳米线的过程中,引入一个温度梯度可调控生长方向。温度梯度驱动前驱体热扩散至温度较低的催化剂界面,聚集并加速催化分解,提升液相化学势,扰动界面稳定性和成核动力学对称性,从而控制纳米线生长方向(图13)。在集成电路中常见的焊点不单是物理过程所形成,在焊接过程中其实存在着显著的温度梯度和形成界面金属化合物的化学过程。有研究组利用同步辐射图像细致观测了在温度梯度作用下随时间在Cu/Sn-9Zn/Cu焊点冷-热两端界面处形成界面金属化合物的变化过程。在冷端形成明显的柱状Cu5Zn8化合物后,由于Zn元素随温度梯度进行热扩散后浓度降低,继而产生Cu6(Sn,Zn)5化合物。

图13 温度梯度导致的硅纳米线生长过程中出现的弯折TEM图
光与物质相互作用的研究始于爱因斯坦对光电效应的揭示,奠定了光化学与光谱学的基础。分子与特定光模之间的相互作用取决于分子的电子结构和振动特征。传统上,光谱和光化学研究假设入射的交变电磁场不受分子或附近除了介电环境外其他因素的影响。然而,已有研究表明环境对入射电磁波的响应显著改变分子所感受到的场效应,受周围物质结构和性质明显影响。光-物质强耦合会达到1 eV,显著影响化学反应动力学,尤其在有机分子体系中,通过形成极化激元以及多分子相干作用导致集体效应(Collective effects),调控反应进程。如图14所示,光-物质耦合可诱导违反经典的Hund规则的单线态和三线态能量反转,操控分子体系中的能量流动。强耦合不仅改变激发态能量,还扰动基态平衡,进而影响分子反应速率、电导率、激发态弛豫路径,为无需改变分子结构的性质调控提供了新途径。
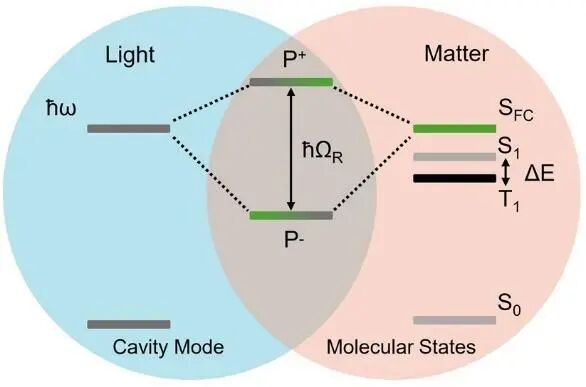
图14 光-物质强耦合状态下第一单重态S1和三重态T1能量反转的Jablonski图,光在光学空腔内与有机分子的Franck-Condon态SFC产生强耦合,形成了杂化的P+和P-光-物质态
在强电磁波辐照条件下(如宇宙射线或核乏燃料处理),化学反应涉及复杂的电子-电子、电子-原子核、原子核-原子核的碰撞,表现出与常规光化学显著不同的行为。需要强调的是,化学反应通常受多种外场(应力场、光场、电场等)的耦合调控,而非单一外场主导。这种多场协同作用显著增强了对反应动力学和选择性的调控能力,为凝聚态化学研究提供了新的理论与实践方向。
2.4 微环境
现有的研究已表明反应位点在反应过程中的动态结构演化会受到其周围微环境的显著影响。尽管微环境中的物质结构未必直接参与化学反应,但其动态响应间接调控了反应位点的行为,进而影响到化学反应过程。本文将反应位点附近不直接参与反应但影响反应动力学与选择性的因素统称为“微环境”。受篇幅限制,本节仅对集中典型微环境因素(局部内建电场、pH值分布、介电屏蔽、空间限域)进行简要分析,以强调微环境在化学反应中的重要作用。
除了外电场外,通过局部结构设计(如杂原子掺杂、官能团修饰或空位缺陷创建)可诱导电荷重新分布,形成短程内建电场。与外电场不同,内建电场作用范围较小,通常沿着反应轴向或键轴的方向施加作用,显著影响体系的能带结构、费米能级、载流子类型以及传输速率,优化反应物种的吸附/脱附等过程,并降低反应能垒。除此之外,内建电场可调控反应位点附近的物种的浓度和种类,促进传质并引发微环境因素的耦合效应。然而,不恰当的内建电场可能阻碍反应进程。早期内建电场研究主要集中于半导体异质结界面物理特性,而在化学领域,其应用多见于离子电池、太阳能电池、光催化及电催化体系,主要用于调节能带结构和增强载流子分离。例如,在ZnO/Ni(OH)2异质结光阳极中,压电效应诱导的内建电场显著影响析氧反应效率。0.2%的拉伸应变会增加约85%的电流密度,而0.2%的压缩应变则会降低约30%的电流密度,反映了内建电场方向对反应效率的差异化影响。在电催化反应体系中,有实验观测到,CuCl(111)/rutile-TiO2(110)异质结内建电场会导致NO3-在界面富集,将超低浓度的硝酸盐高效转化为氨,并通过内建电场提高中间体*NO能量降低决速步能垒,进而提高反应转化效率,使产氨选择性接近99%。
局部应变、缺陷、杂原子、官能团吸附等引发的电荷动态排布可改变反应位点附近物种分布,导致局部pH值波动。pH值调控是操控化学反应过程的常规手段,但反应位点附近随反应进程动态变化的pH值对化学反应的影响更为复杂。从微观角度看,反应过程中对酸性或碱性物质的消耗会引起局部pH值的瞬时波动。在光催化或电催化二氧化碳还原(CO2R)和水分解产氢反应中,阳极产生OH-导致光催化剂或电极附近的pH值升高。根据方程Nernst方程,CO2R和H2O分解全反应中氧化还原电势与pH值之间呈线性关系。电催化反应速率更快,pH值波动更加明显,尤其是电催化CO2R中,实验证实,电极表面局部pH值直接影响反应的选择性。早期实验研究认为高pH值更利于双碳产物的生成,而近期的研究表明较低pH值(H+离子浓度升高)增加阴极附近CO2浓度,促进CO或双碳产物(如C2H4、C2H5OH)生成。这些相悖的实验结果充分表明了局部pH值在CO2R反应中的复杂作用,亟需在充分考虑微环境因素下进行深入研究。
微环境中的局部电场或物种浓度变化显著影响反应位点附近的介电环境,尤其在光催化反应中作用突出。研究表明,半导体材料厚度直接影响光学性质。例如,CdS纳米薄膜厚度从15.2 nm增长到25.4 nm,其光学带隙从2.88 eV降低到2.61 eV。多体微扰理论揭示,离域介电屏蔽效应对低维半导材料电子结构影响显著。层状材料随厚度减小,准粒子带隙显著增加。例如,4层黑磷的准粒子带隙约为0.63 eV,3层体系则增加到0.87 eV,2层为1.22 eV,而单层黑磷的准粒子带隙已经增加到了2.08 eV。介电环境的还影响激子的结合能,单层材料的激子结合能通常都会显著大于其体相材料。在设计二维Janus材料(Ga2SO或InSO0.5)时,材料自身厚度以及衬底材料(如宽带隙h-BN)厚度均可调控激子结合能,幅度大(200~250 meV),结果如图15所示。实验上,通过介电环境设计,单层WS2和WSe2材料的带隙和激子结合能可在数百meV范围内调控。
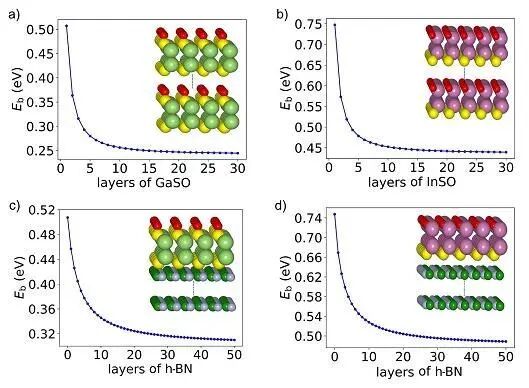
图15 (a) Ga2SO与(b) InSO0.5层数与其激子结合能之间的关系;(c) 单层Ga2SO与(d) 单层InSO0.5置于h-BN之上后其内部激子结合能受衬底材料厚度的影响
晶体的生长过程受到周围环境的空间限域显著影响,特别是在多孔体系(如生物体或多孔矿物)的孔道中。仿生研究通过调控离子组成、pH值以及过饱和度等因素控制晶体的形态和强度。纳米约束空间中的晶体生长动力学受限,离子在液膜内的二维质量传输受到空间梯度控制,影响晶体的表面形态和生长速率,表现为倾斜螺旋或局部化成核等特征。分子动力学模拟表明,NaCl过饱和溶液中, Na+在亚稳态NaCl(001)/水相界面选择性吸附,溶剂的极化会显著影响极性界面离子分布,进而调控晶体生长。
3 凝聚态物理新概念驱动的化学新方法
化学与物理学在研究视角与方法上存在根本差异。尽管凝聚态化学研究无需严格追随凝聚态物理学的进展,但新兴的物理学概念和现象为化学发展提供了新思路。本节介绍若干较新的物理学概念,这些概念已经在化学研究当中展现出应用潜力。需要强调的是,化学研究的核心在于反应,将凝聚态物理学概念应用于化学反应是凝聚态化学的重要方向。由于物理学新现象根源于物质结构,探索其在化学反应中的作用是凝聚态结构化学的关键任务之一。
3.1 量子限域效应
量子限域效应源于量子力学,描述当材料的尺寸趋近或者小于de Broglie波长(物质波波长)时,电子或电子-空穴对(激子)的能量发生量子化,局限于离散能级,导致能谱呈非连续分布。这一现象是纳米材料展现不同于体相材料物理性质的根本原因。20世纪90年代,纳米金属或半导体团簇因离域价电子的独特性质引起化学家的关注。在化学研究当中,即便是对于尺寸较大的体系,表面原子与体相原子因配位数差异表现出不同特性,随尺寸减小及空间维度降低,微小的几何构型变化也会显著影响电子性质及环境响应,进而调控化学反应进程。需要指出的是,化学反应不仅受量子限域效应影响,还涉及空间限域对反应物浓度和构型、分布姿态、局部有效压力以及热传导等因素的综合调控。本节仅聚焦量子限域效应对化学反应的影响。从量子力学视角,电子波函数受限于狭小空间时,其电荷在电磁场中的非线性位移显显著增强。限域空间不仅包括材料整体尺寸,还涵盖体相或表界面的缺陷与杂质。量子限域效应在化学的应用包括在低维催化材料、团簇波函数叠加导致的共振和间接隧穿效应引起的自组装反应、缺陷工程设计的催化剂,以及过渡金属原子分散在基底材料表面形成的单原子催化剂等。
3.2 表面等离激元
为便于理解,首先澄清相关概念。等离子体(Plasma)也被称为固态、液态、气态之外的第4种物质状态,是整体保持电中性的电离物质系统,包含了阳离子、阴离子、中性粒子以及电子,具有高导电性,对电磁场响应强烈。尽管传统等离子体多位气态,但其集体波动特性使其纳入凝聚态物理学研究范畴。随着相关研究的深入,受电磁场驱动的固体或液体中的自由电子体系亦可视为等离子体。等离子体的高能量状态赋予其化学高反应活性,可于固体表面、液体或微生物发生相互作用。广泛应用于表面刻蚀、表面改性、缺陷工程等。等离子体激元或等离激元(Plasmon)是等离子体共振的量子化形式,具有长程的电子-电子相关性,研究主要集中于金属纳米粒子、金属表面以及金属-半导体界面。等离激元可理解为金属中阳离子周围电子密度周期性振荡的量子化。当等离激元与光子发生耦合时,形成等离激元极化子(Plasmon polariton)。低于等离激元频率的光在材料表面发生反射,而高于这个频率的光则因电子响应不足而在材料中传播。当频率匹配时,在金属-空气或金属-半导体界面形成表面等离激元极化子(Surface plasmon polaritons),电磁波以近红外或可见光频率沿界面传播,其中在金属表面出现的电子的相干振动被称为表面等离激元(Surface plasmon),而在空气或半导体上传播的电磁波被称为极化子(Polariton)。表面等离激元极化子局限于界面,引发强烈的光-物质相互作用,传统应用于光学传感、显微以及通信等领域。近期,局域表面等离激元共振(Localized surface plasmon resonances)在化学反应中的应用备受关注,因其诱导金属表面明显的电荷分离以及近场振幅增强,对化学反应过程产生重要影响。如图16a所示,在特定波长的光照条件下,金属纳米粒子表面会由局域表面等离激元共振效应产生局部电场。金属纳米粒子表面等离激元共振主要会产生3种效应:(1)电磁场的增强效应;(2)载流子效应;(3)热效应。这3个效应的时间尺度按顺序从飞秒逐渐增加到纳秒范围。金属纳米可以与半导体材料表面靠近时,可以在其界面处观测到被增强的电场,并产生多因素耦合效应,如图16b所示。金属团簇的几何形状会显著影响其共振所需入射光波长和局域表面等离激元的分布。如图16c中的理论计算结果所示,电场会倾向于集中在纳米颗粒尖锐的顶端,并沿着直线边缘迅速衰减。团簇间的距离调控,显著影响电场增强幅度以及峰值波长。此外,局域表面等离激元共振可以提供多重催化效应,包括分子内电荷转移、热载流子转移以及热驱动作用。图16d中展示的是Ag20纳米粒子局域表面等离激元共振对CO2分子中C—O键断裂的影响。通过图16e可以看出,虽然Ag20纳米粒子在CO2轴向提供了一个被增强的电场强度,但是在CO2附近位置已经出现了极大的衰减,并不能直接导致C—O键断裂的程度。通过对CO2分子上电荷随时间的变化进行观察,如图16f所示,可以发现在16 fs时CO2分子从Ag20纳米粒子上得到了0.63个电子,从而说明这个Ag20纳米粒子光催化还原CO2反应是由局域等离激元共振带来的电荷转移导致的。这是表面等离激元作为凝聚态物理学概念在化学催化反应中的典型应用。
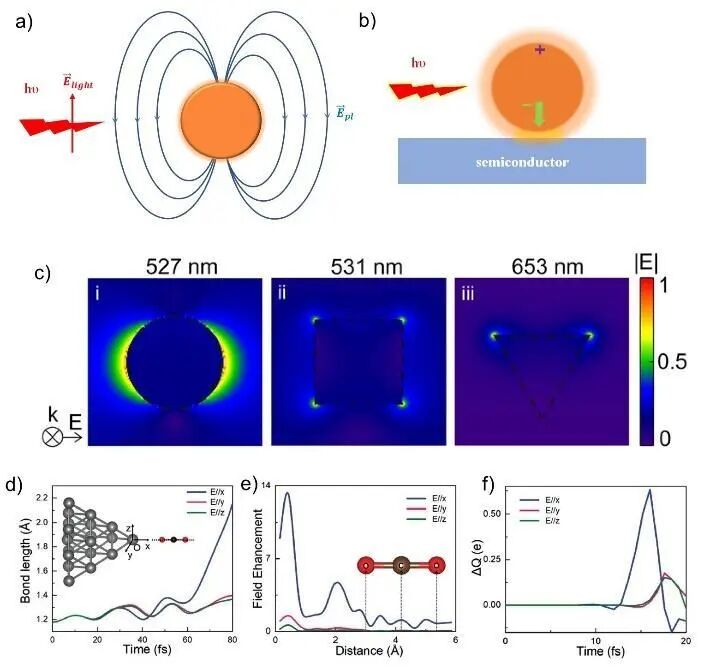
图16 (a) 光激发金属纳米团簇表面等离激元共振电场方向示意图; (b) 半导体晶体表面支撑的金属纳米团簇在光激发情况下表面等离激元效应耦合效果示意图;(c) 时域有限差分法(FDTD)计算不同几何形状金属团簇上局域表面等离激元共振归一化电场分布图;(d) 基于Ehrenfest方案执行的非绝热动力学模拟局域表面等离激元增强Ag20团簇催化CO2R过程中C—O键长随时间的变化;(e) Ag20团簇表面等离激元对入射光电场强度的增强;(f) 局域表面等离激元引起CO2分子上电荷随时间变化
3.3 拓扑绝缘体
根据能带结构,物质通常分为:绝缘体、金属、半导体、半金属(Semi-metal and half metal)。由于绝缘体和半导体能带间无明确界限,二者统称为绝缘体。拓扑绝缘体(Topological insulator)是一类体相表现为绝缘体,却具有导电边缘的材料体系,其表面态与普通绝缘体显著不同(图17)。3D拓扑绝缘体具有导电表面态,2D拓扑绝缘体则具有1D线状导电边缘态。相对于普通的绝缘体来说,拓扑绝缘体的能带呈现“扭曲打结”特征,无法通过连续的局部微扰将拓扑绝缘体转变为普通绝缘体,除非解开电子波函数相互扭曲形成的拓扑结。拓扑绝缘体在相图中独立于普通绝缘体,二者之间由导电相连接起来,并且不能用Landau对称破缺理论进行描述。目前,拓扑绝缘体的研究主要集中在物理领域,潜在应用于自旋电子器件、无耗散晶体管、磁-光电器件等。在化学反应中的应用尚少,多利用其组分中的过渡金属,而非拓扑绝缘体的特性。近期,日本学者报道以硒化铋(Bi2Se3)纳米颗粒催化胺与一氧化碳和氧气进行氧化羰基化合成尿素衍生物,在20 ℃条件下完成丁胺的羰基化,4 h内产率高达99%,如图18a所示。为验证拓扑特性作用,研究人员测试了不同催化剂催化效率。图18b显示,块体的Bi2Se3催化丁胺羰基化反应的周转频率(Turnover frequency, TOF)非常高,但产物形成速率较低。因为块体催化剂比表面积小,暴露在表面的催化位点有限。当使用Bi2Se3纳米粒子作为催化剂后,TOF保持稳定,而产物的形成速率显著增加,凸显比表面积的关键作用。随后,将催化剂内的Bi元素分别用其他元素进行替换后发现,TOF都出现了显著的下降,说明单独Bi和Se元素对催化反应起不到主导作用。综合上面的结果可以发现,对这个催化反应起到最主要作用的是Bi与Se的协同作用,或者说是Bi2Se3的表面态。理论计算结果表明,拓扑绝缘体的表面态将三重态的氧分子转化为单重态,促进氧分子的活化以及尿素衍生物生成,验证了拓扑表面态的催化贡献。该研究表明,Bi2Se3所展现的拓扑绝缘体性质可以在较温和的条件下催化类似的氧化羰基化反应,合成处多种尿素衍生物。展望未来,拓扑绝缘体表面导电态对半导体对载流子的传输和分离作用有望在化学反应中获得更广泛应用。此外,拓扑材料可扩展至拓扑半金属(Topological semimetals),包括Weyl半金属、Dirac半金属、结线半金属(Nodal-line semimetal)和多代结点半金属(Multidegenerate nodal-point semimetal),其化学应用潜力尚待探索。
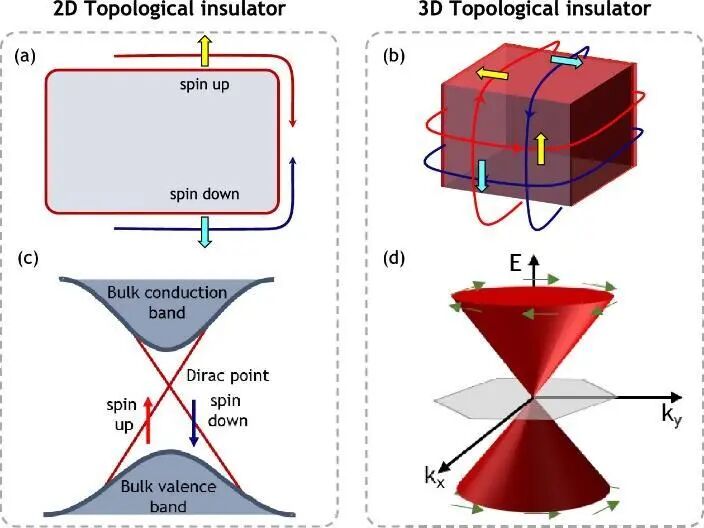
图17 (a, b) 二维和三维拓扑绝缘体的表面导电态示意图;(c, d) 相应能带结构示意图
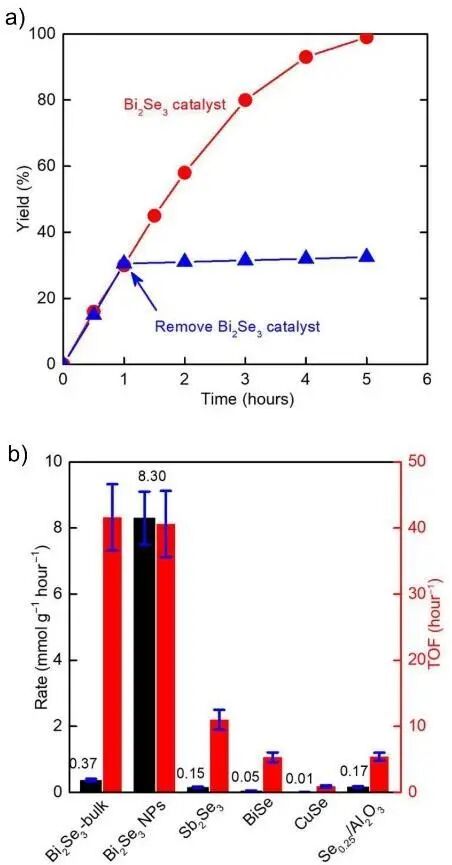
图18 (a) 拓扑绝缘体Bi2Se3催化合成1,3-二丁脲的产率;(b) 不同催化剂形态和组分对产物形成速度和催化转化率的影响
结论与展望
本文简单介绍了凝聚态物理的发展历程以及这个学科名字的变化,认识到凝聚态物理学与凝聚态化学在研究领域上的主要区别。通过对典型的固-气、固-液、固-固界面反应中的结构动态演化,强调了凝聚态结构化学的核心:反应条件下,物质体系结构持续动态演化,化学研究需充分考虑相关因素的影响。外界条件(如力、电、磁、光场)协同作用引发物质结构变化,驱动化学反应进程,并伴随持续的动态结构演变。这些结构演化进一步影响反应位点附近微环境(如局部电场、pH值、介电屏蔽、空间限域),从而调控反应动力学与选择性。
然而,受限于当前计算能力,量子力学理论在复杂化学反应体系中的应用仍存在不足,难以全面还原真实反应过程。由于化学反应体系的复杂性和多样性,尚无统一的理论框架能够描述绝大多数化学反应。随着凝聚态物理学研究的深入,新型物质现象不断涌现,为化学领域提供了新的研究视角与应用潜力。例如,量子限域效应、表面等离激元及拓扑绝缘体等物理概念已在催化、光化学及材料合成中展现出重要价值。这些进展为大力发展凝聚态化学及凝聚态结构化学提供了坚实的理论与实践基础。
作为重要实验表征手段,超高时空分辨率原位表征技术(Ultrafast time-resolved in-situ techniques)在可见的未来会得到长足的发展,包括且不限于:超快瞬态吸收光谱、原位超分辨显微镜、时空组学技术、超短激光脉冲扫描隧道显微镜等。这些原位表征技术可以在真实反应条件下提供飞秒和亚纳米尺度的实时观测数据,从而为凝聚态化学研究提供第一手的实验观测数据和相关重要证据。随着人工智能技术的快速进步和迭代,研究人员有望通过人工智能或机器学习的辅助来研究更复杂的凝聚态化学过程,进行更加细致的动态结构模拟,提供更加贴近现实的凝聚态化学理论研究结果。先进的原位表征和人工智能等技术的结合将为化学研究的进步提供强劲的推动力,令化学科学真正进入凝聚态的范畴,为人类对化学反应机理的解析、催化剂的设计、多场耦合调控、高效节能的进行化学合成等方面取得更大突破提供必要的科学前置条件。跨学科融合,特别是凝聚态物理学与化学的协同,将进一步推动新型化学反应路径的发现及应用,为能源、环境及材料科学等领域提供创新解决方案。
推文篇幅有限,欢迎阅读原文,共飨学术
Authors:Ran Jia, Jian Wang, and Wenfu Yan*
Title:From Condensed Matter Physics to Condensed Matter Chemistry
Published in:Progress in Chemistry, 2026,38(1): 1-22
DOI:10.7536/PC20250626

 概况
概况