近日,李辉教授课题组在复杂体系分子振动研究方面取得重要进展。相关成果发表于杂志Journal of Chemical Theory and Computation,文章题目为“Machine Learning Approach Based on a Range-Corrected Deep Potential Model for Efficient Vibrational Frequency Computation”[1]。该工作作者为吉林大学化学学院、理论化学研究所杨积泰、丛阳、李由和李辉。
研究背景:
光谱模拟是连接分子动力学与光谱实验观测的桥梁。通常光谱模拟的技术路线有两种:一是计算轨迹中体系整体各时刻的电偶极矩和极化率等物理量,之后计算相关函数得到光谱。该技术路线也可以借助DeePMD-kit的功能(红外/拉曼光谱计算工作流@Notebook,快速上手光谱表征预测和计算);二是从单个生色分子/生色团出发,计算轨迹中生色分子/生色团各时刻的振动频率,进行统计得到光谱。
后者从单个生色分子/生色团及其局域环境开始计算,原子数很少,使得研究者可以采用更高精度的方法。另外,计算分子的瞬时振动频率可帮助分析氢键、相对位置、静电场、分子内变形等各种效应与频率的关系。因此后者是李辉研究组主要关注的技术路线。研究组开发了局域量子嵌入框架[2],该框架的特点是高精度的量子方法处理分子振动,再将得到的量子力学部分嵌入动力学轨迹当中。如此得到的瞬时振动频率既包含振动的量子效应,又包含局域环境效应。然而,尽管目前该框架的计算成本尚可承受,当动力学轨迹长度达到纳秒量级时,计算量会变得相当可观,在多维振子问题当中,该技术路线的计算成本也会非线性增长。
与机器学习方法结合:
该框架的计算成本主要在瞬时频率计算。从体系坐标到频率的映射是一个复杂高维函数,且频率计算在流程中会重复几万甚至十几万次。可以将轨迹中的坐标和高精度方法得到的振动频率值制成训练集,再利用机器学习方法,训练出能将分子坐标映射成振动频率的函数。这个思路与借助机器学习进行势能计算相同,只是将计算目标从势能变为了振动频率。
研究组希望最后的计算流程简单实用,使用者不需要太多与机器学习相关的知识,并且能轻松地利用其他研究者的数据集,因此选择具有良好社区支持和接口支持的DeePMD-kit软件来帮助振动频率计算。
结果表明,常用的DP(Deep Potential)模型可以达到光谱模拟要求的精度,大大增加计算效率,但是训练时间很长。研究组注意到,DP模型的变体DPRc(Range-Corrected Deep Potential)模型可能更适用于振动频率的计算。DPRc模型为了针对QM/MM方法得到的能量进行修正,将重点放在QM-QM区域与QM-MM区域原子间相互作用上,而不包含MM-MM区域原子间相互作用。与之相似,振动频率是分子本身的性质,主要受局域环境原子的影响,忽略非局域环境原子的原子间相互作用,理论上对结果影响很小,且能很大程度减少模型所需的参数,提升训练效率。在DPRc模型中,研究组将原QM对应的区域设为与生色分子相关的区域,而原MM对应的区域包含其余的原子。

DPRc模型区域划分示意图:以甲酸溶液为例,甲酸分子被设为对应的QM区域,其余溶剂水被设为对应的MM区域。
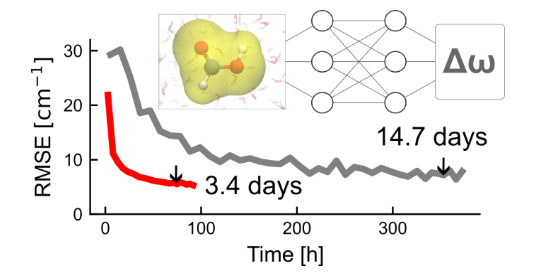
结果证明了我们的想法,这种策略大大节省了训练时间,而且对精度影响很小,DPRc在精度、计算效率、训练时间上都达到了光谱模拟的实际需求。我们同时也对需要的数据集大小进行了测试,发现仅使用一千个左右数据点做为训练集,也可以达到满意的精度。

乙腈溶液中CN伸缩振动频率计算精度在不同大小训练集下的变化。
总结:
该研究将局域量子嵌入框架与DPRc模型结合,验证了关注局域环境的计算思路,大大提升了光谱模拟的振动频率计算效率。该工作可以协助长时间、复杂的光谱模拟计算,如多维振动、二维光谱模拟、和频光谱模拟等。
课题组网页:
http://huiligroup.org/zh/index.html
参考文献:
[1] Yang, J.;Cong, Y.; Li, Y.; Li, H., Machine Learning Approach Based on a Range-Corrected Deep Potential Model for Efficient Vibrational Frequency Computation. Journal of Chemical Theory and Computation 2023, 19 (18), 6366-6374.
[2] Cong, Y.;Zhai, Y.; Yang, J.; Grofe, A.; Gao, J.; Li, H., Quantum vibration perturbation approach with polyatomic probe in simulating infrared spectra. Physical Chemistry Chemical Physics 2022, 24 (2), 1174-1182.

 概况
概况