近日,吉林大学化学学院刘靖尧教授团队在电催化CO2还原制备多碳产物的新策略研究方面取得重要进展。受自然界中多酶串联催化机制的启发,研究团队创新设计了双位点单原子纳米酶,并借助巨正则系综下的恒电势密度泛函理论(GCE-DFT)系统揭示了串联位点间的空间分离反应路径,有效提升了乙醇产物的选择性。在此基础上,团队进一步引入脉冲电催化策略,通过对关键反应步骤的时间维度进行精细调控,实现了乙醇生成路径在热力学和动力学上的双重优化。该策略为实现高效CO2电还原制备多碳产物提供了新思路与理论支撑。相关研究成果以题为 “A Pulsed Tandem Electrocatalysis Strategy for CO2 Reduction” 的论文发表在J. Am. Chem. Soc. 上 (JACS, 2025, 147(17), 14388-14400)。
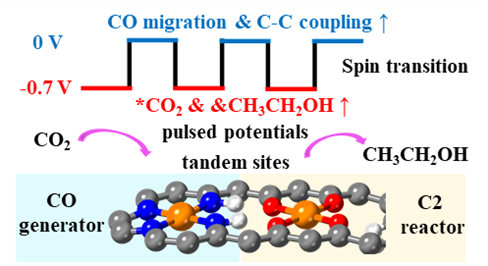
图1. 脉冲串联电催化策略示意图。
在前期研究中,研究团队基于仿酶催化原理,构建了一系列单原子纳米酶,并借助机器学习特征工程提出了基于电子结构的催化性能描述符,高效筛选出多种CO2还原反应(CO2RR)潜在催化剂(ACS Catal. 2024, 14, 14021)。在此基础上,团队进一步模拟乙烯形成酶中FeO4活性中心的特征,构建出含FeN4与FeO4双位点的串联催化剂体系(图1),实现了CO2活化、CO生成与C–C偶联的空间分工,并在–0.7 V(vs. SHE)下实现乙醇的高效生成。进一步分析表明,CO在FeN4位点生成后易于向FeO4位点迁移,随后在较低能垒下与第二个CO分子发生C–C偶联形成C2产物(图2)。然而,进一步计算显示该过程的能垒随电位变负而上升,为引入电位调控策略提供可能。研究团队由此提出脉冲电催化策略,在–0.7 V与0 V之间周期切换,精准调控关键步骤热力学与动力学:在0 V时,CO迁移和C–C偶联的能垒分别从0.25 eV和0.62 eV降至0.05 eV和0.09 eV,并抑制中间体过度还原;而–0.7 V则有利于CO2吸附与关键中间体(如*CH3CH2OH)形成。该策略显著提升了乙醇产率与选择性。
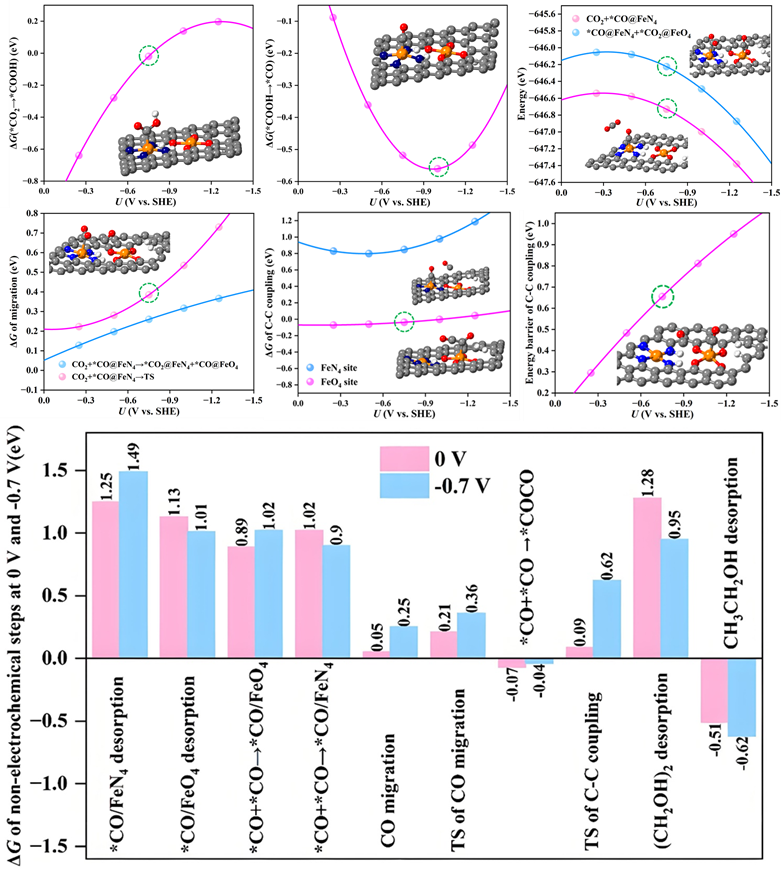
图2. FeN4-FeO4上关键步骤能量变化及脉冲电催化分析。
该策略亦被拓展至Mn、Co、Ni、Cu、Zn等多个金属体系。其中CoN4-CoO4双位点催化剂在–0.8 V和0 V脉冲切换下表现出良好的C2产物选择性,验证了其结构-电位协同策略的普适性。
为进一步阐明脉冲调控机制,研究团队对FeN4与FeO4在不同电位下的电子结构进行了分析。结果表明,FeO4位点在由–0.7 V切换至0 V时,自旋态由低自旋(S=1)转变为高自旋(S=2),伴随着自旋密度与磁矩的同步变化(图3)。该自旋态演化有效促进了CO迁移与C–C偶联,为脉冲催化的电子电子本质提供了理论支撑。
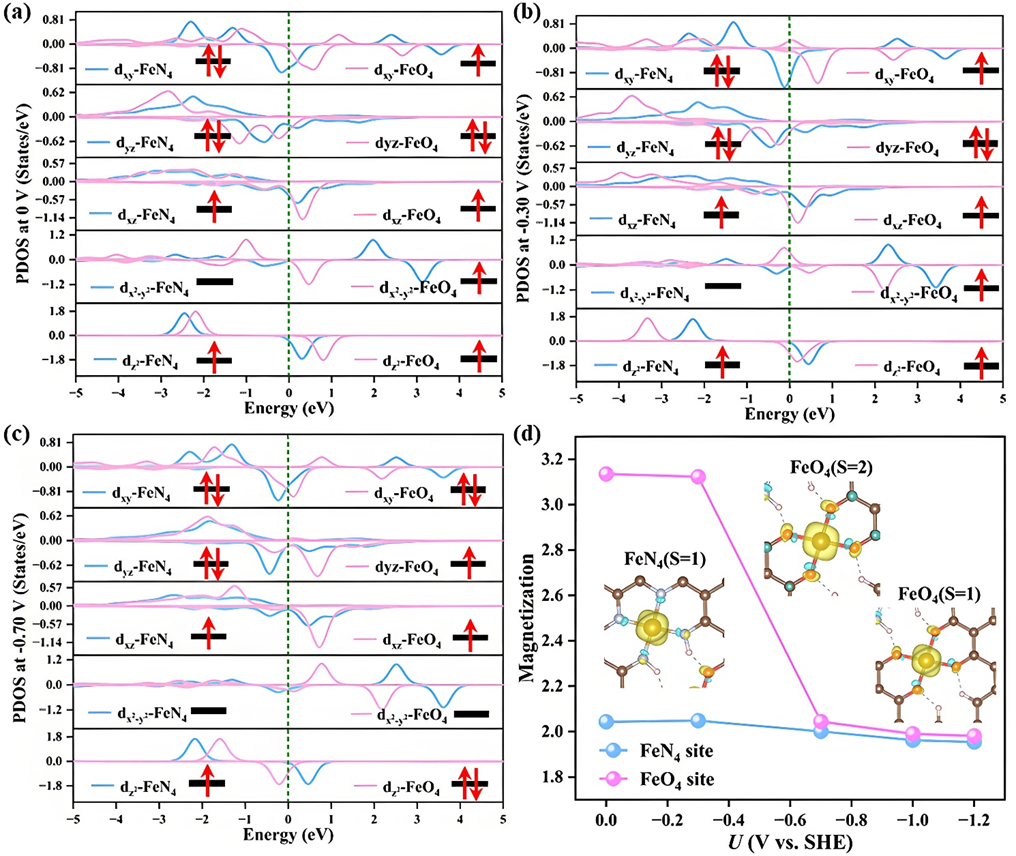
图3. 不同电位下活性位点自旋态和磁矩的演变情况。
该研究工作创新性地将“空间分离的串联位点结构”与“电位脉冲时间调控策略”相结合,构建了一个可实现CO2还原反应关键路径“时–空分离”调控的新范式,在提升C2产物生成效率与选择性的同时,为动态电催化系统的设计与机理研究提供了理论依据与方法指导。
吉林大学化学学院博士研究生孙昊为第一作者,刘靖尧教授为通讯作者。该研究得到了科技部重点研发计划等项目的经费支持。
课题组网站:
https://www.x-mol.com/groups/liu_jingyao
论文链接:
https://pubs.acs.org/doi/10.1021/jacs.5c00633

 概况
概况